
Cutaneous mosaicism
KEY POINTS
-
The field of mosaicism has changed substantially in the last decade
-
Mosaicism is defined as the coexistence of at least two genotypes in an individual derived from a single zygote, by the time of birth, and which gives rise to a disease phenotyp1
- Mosaic disorders can therefore not be inherited, but increasing numbers of them have been found to have the potential to be passed on to future offspring - if passed on they will be passed on as a germline disorder, not a mosaic disorder
- Mosaicism affecting the skin leads to recurrent developmental patterns of skin disease, but these patterns are neither exhaustive nor exclusive, as they can also be seen in germline genetic conditions. Genotyping is therefore essential for directing genetic counselling
- Genotyping is increasingly important for directing targeted therapies
Autor: Prof. Veronica Kinsler, London
Introduction
As physicians working in the field of Dermatology we are often proud of the clinical phenotyping skills which separate us from other specialties. The ability to make a difficult diagnosis at a single glance is not only impressive – indeed it was what first attracted me into the specialty of Paediatric Dermatology - but is a badge of experience earned through hard work. Despite the importance of these skills, in the field of mosaic disorders it is irrefutable that the recent discoveries in molecular genetics have disproved many facts we thought we knew, and overturned hypotheses we thought we understood. We have now arrived at a confluence of phenotyping and genotyping which has enabled us to redefine mosaic disorders to some degree, and to understand the clinical relevance of getting that definition right. This article will attempt to impart that new understanding.
The study of mosaicism at genetic level started in experiments on Drosophila in the early 20th century, and the first proofs of the disease mechanism in human skin were published in the 1990s. These were discovered using germline conditions to generate a candidate gene approach. Mosaic KRT10 mutations* were thus found in parents with an epidermal naevus as a result of familial transmission of epidermolytic ichthyosis to their children.2 GNAS mutations in skin lesions in McCune-Albright syndrome3, were found after genetic studies in Albright’s hereditary osteodystrophy. Further discovery in mosaics was then delayed until the last decade, as without candidate genes Sanger sequencing was not sufficiently sensitive to detect mosaic mutations reliably.
*It has become fashionable to use the term “variant” rather than the term “mutation”, due to the need to differentiate between variants which are pathogenic, of unknown significance, and benign. Where I have used the term mutation this is because these variants are known to be pathogenic.
Definitions and terminology
Recent studies have discovered a surprisingly high rate of spontaneous mutations occurring during normal embryonic/fetal development4, and it is also known that post-natal somatic mutations in normal and tumour tissues are so common as to be effectively universal over time5. If we were to continue with the original definitions of mosaicism – two genotypes in an organism derived from a single zygote - then everyone in the world would now be considered mosaic. In order to define mosaicism as a disease state, a recent consensus from the European Reference Network (ERN) proposed the following:
“Mosaicism is the coexistence of at least two genotypes in an individual derived from a single zygote,
by the time of birth, and which gives rise to a disease phenotype"1
This therefore excludes any non-pathogenic variants which happen before birth, and any somatic variants which arise after birth.
A germline mutation is one which affects the whole individual and can either be inherited or can arise de novo at zygote stage. A post-zygotic mutation is defined as a mutation which occurs after the single cell stage but before birth and can therefore not be inherited.
Classification of mosaic disorders
Classification of mosaic disorders has also been subject to change recently, partly due to new genetic knowledge, and partly driven by the need to find a clinically-meaningful system. One traditional classification divided mosaicism into somatic (non-gamete tissues only), gonadal (gametes only), and somatic with gonadal. Whilst this is theoretically useful clinically, in practice it is usually not possible to determine to which class an individual patient belongs, as it requires genetic testing of their gametes, which even in males is rarely done.
We therefore turn to classification based on genetic mechanism. This was first started by Happle, who suggested “type 1 and type 2 segmental mosaicism” for autosomal dominant mutations.6 More recently, a European consensus paper has built on this to produce a larger framework (Table 1). This permits an understanding of risk of transmission to future children. In addition, it builds a framework into which future cutaneous mosaicisms should be able to be incorporated.1
There is an exception to this, which is that it does not allow for mosaic disorders which are only caused by two somatic mutations, either in the same or different genes, and if in the same gene on the same or different alleles. These could theoretically arise on any germline background. Known examples of this are double cis TEK mutations causing Blue Rubber Bleb syndrome7, and congenital plexiform neurofibromas in patients without germline NF1 mutations. In these cases, it is important to exclude low level constitutive mosaicism of one of the mutations as a predisposing event.
This new classification requires consideration of the germline genome of the patient – divided into normal, autosomal dominant, or autosomal recessive1 (Table 1). On those backgrounds, a post-zygotic disease-causing mutation occurs, leading to a developmental pattern on the skin at birth or in childhood (very rarely much later in life). With a normal germline genome, the post-zygotic mutation can be either lethal in the germline or not, resulting in no risk to the offspring, or an unknown potential risk to the offspring respectively. Existing knowledge of the mutations and whether they have previously been described in the germline helps allow these outcomes to be predicted. With an autosomal dominant germline genome, a post-zygotic mutation affecting the same gene can lead to an earlier and/or more severe localised phenotype in a developmental pattern. In this instance there is of course a risk of passing on the germline condition to offspring. With an autosomal recessive germline genome, a post-zygotic mutation affecting the same gene can lead to a mosaic phenotype at birth. In this instance there would not be a risk of passing on the condition other than as a carrier.
Table 1 Classification and examples of cutaneous mosaicism based on genetic mechanism, which allows prediction of whether the individual has a risk of passing on disease to offspring in the germline (adapted from 1).
|
Germline genome |
“Normal” |
“Normal” |
Autosomal dominant mutation |
Autosomal recessive mutation |
|---|---|---|---|---|
|
Post-zygotic mutation |
Lethal in the germline |
Compatible with life in the germline |
Second mutation in the same gene (homozygous or compound heterozygous) or theoretically in a different gene with similar effects (dizygous) |
Second mutation in the same gene (homozygous or compound heterozygous) or theoretically in a different gene with similar effects (dizygous) |
|
Example |
Proteus syndrome – mosaic mutation in gene AKT1 |
Segmental/localized mosaic neurofibromatosis type 1 – mosaic mutation in gene NF1 |
Germline neurofibromatosis type 1 with congenital plexiform neurofibroma – germline mutation in gene NF1 with second mutation in NF1 in the plexiform |
Mosaic ectodermal dysplasia with epidermal fragility – germline mutation in gene PKP1 with a second mutation in PKP1 in the affected skin 8 |
|
Offspring at increased risk of germline disease |
No |
Yes |
Yes |
No |
|
Estimated comparative incidence, but these estimations may change with further discoveries |
Most common as a group |
Common as a group |
Rare as a group |
Very rare as a group |
Developmental patterns on the skin and their clinical relevance
Patterns of birthmarks can only really be classified where the lesions are large or multiple. Over the preceding decades up to seven patterns have been hypothesized by Happle to be related to mosaicism6,9 – Blaschko-linear (narrow bands), Blaschko-linear (broad bands), checkerboard, phylloid (leaf or flower-like); patchy pattern without midline separation, lateralization (similar to checkerboard but one side of the body only), and sash pattern.
From my perspective there are three important points to understand about patterns, all three because of their clinical relevance.
The first is that these patterns should be viewed as developmental patterns rather than mosaic patterns, as they are not always caused by mosaicism. The most obvious example is with true fine and whorled Blaschko’s lines as originally described10, which is seen with many X linked germline disorders such as incontinentia pigmenti, and newly described disorders affecting pigmentation11,12. The distinction between developmental and mosaic patterns matters clinically, rather than being just a question of terminology, because we have to understand the genetic mechanisms of what we are looking at before we can manage the condition correctly. More specifically, it matters because of the implications for genetic counselling.
The second is that the designation of what constitutes a separate pattern has not previously been clinically relevant - for example there is no evidence that a unilateral developmental pattern of a mosaic disorder means anything different clinically from a bilateral pattern. What might matter however is the skin cell lineage of origin affected by the mutation. The reason for this is that different cell lineages will have different potential extra-cutaneous associations because of their embryological origins. For example, if we can establish that a fine and whorled blaschkolinear pattern on the skin is caused by a mutation in a keratinocytic lineage, it is potentially going to be associated with other ectodermal issues, but less likely to be associated with neurological abnormalities than for example a mutation in a melanocytic lineage. One example of where there is proof of this concept is from the pattern of facial portwine stains and the likelihood of the associated ophthalmological and neurological complications which constitute Sturge-Weber syndrome. Portwine stains affecting the forehead reflect an embryonic vascular lineage which derives from the developing forebrain, giving rise to the frontotemporal facial placode, in addition to the vasculature of the cortex and developing eye, and this is likely to be the reason why this location is so highly predictive of these associated complications13. This proof of concept can therefore potentially be applied to other types of patterns and this assessment across diseases is underway.
The third important point about the original seven patterns or this new three patterns is that neither is exhaustive. These groups sometimes accommodate vascular lesions, but frequently don’t, which may be to do with the rapid remodelling of embryonic vasculature as it develops and the complex array of different blood vessels which result. In addition, they don’t cover patterns caused by mosaicism affecting other cell lineages in the skin such as connective tissue, which so far have resisted classification as so little is known in that area.
In summary, cutaneous mosaicism is not confined to these patterns, and these patterns are not confined to cutaneous mosaicism.
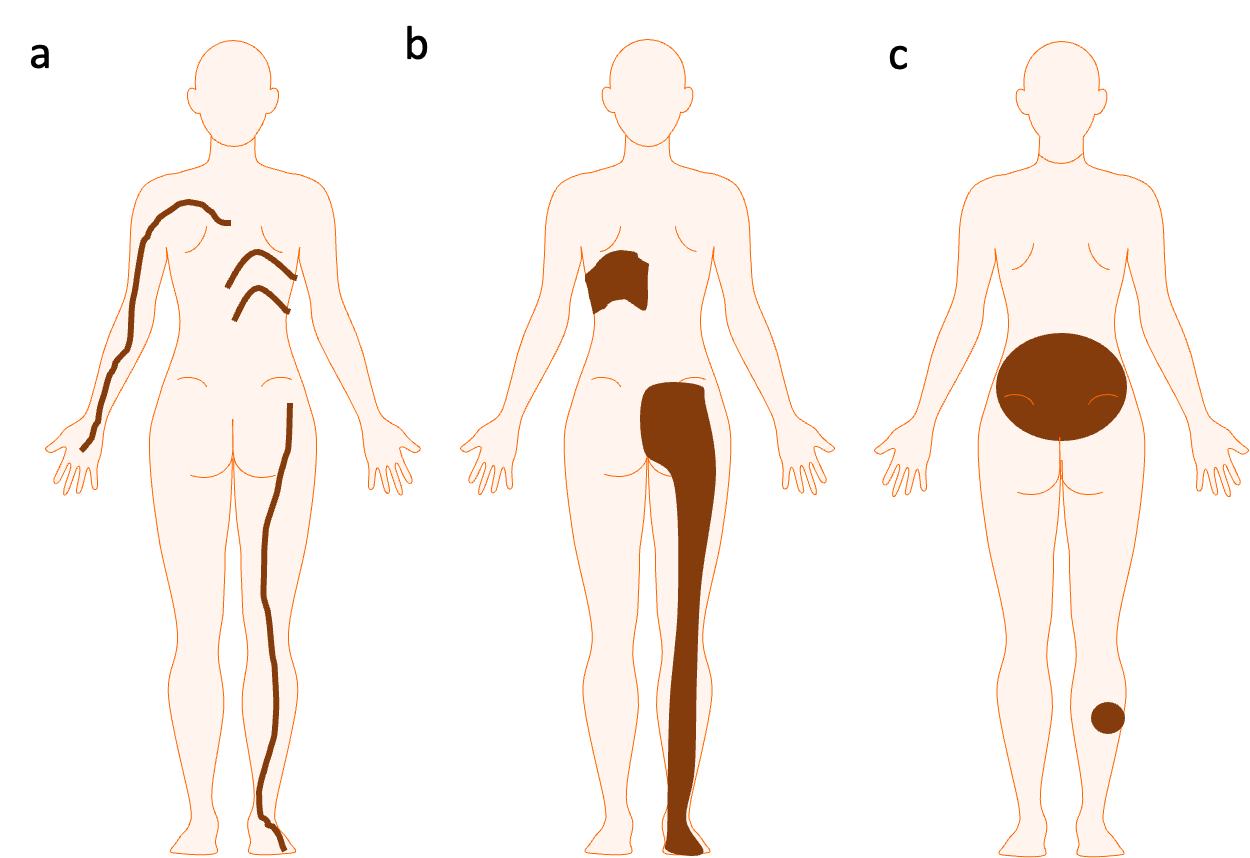
With that in mind the three recurrent patterns (Figure 1) which are lineage-specific and therefore potentially helpful in making a clinical diagnosis and in understanding the potential extra-cutaneous associations of a disease are the following, as established by a large study of patterns of congenital pigmentary disorders14:
- Blaschko-linear pattern – to be called this it should include at least some areas of narrow patterns, i.e. bands of up to approximately 1cm wide in a newborn, which are linear on the limbs and/or whorled on the trunk, and which respect the midline anteriorly and/or posteriorly. This has single or multiple bands, and can be unilateral or bilateral. This is an epidermal lineage cell pattern. It is therefore seen in epidermal naevi, and in hypo- or hyperpigmented Blaschkolinear patterns which are caused by mosaic abnormalities in the epidermal lineages which affect the ability of keratinocytes to handle pigment. It is not seen in any melanocytic or vascular mosaicism.
- Quadrilateral or segmental pattern – this has quadrilateral shapes, superimposed on the body in 3D; single or multiple, and can be unilateral or bilateral. It is characterized by vertical midline cutoffs anteriorly and/or posteriorly, and roughly parallel linear upper/lower edges. This can occupy more or less of a body part, for example relatively small quadrilaterals on the trunk, or a whole body segment such as arm, shoulder and upper torso. This pattern therefore includes the previous terms broad Blaschko-linear, flag-shaped, and checkerboard. This pattern is a melanocytic pattern as it is seen with mosaicism affecting melanocytes (can be increased or decreased pigmentation) and can also be seen with vascular mosaics.
- Circular or non-segmental pattern – this is, round-edged, and lesions can be single or multiple, variable sizes, with or without a dominant clearly larger lesion. Where present the dominant lesion may or may not cross the midline anteriorly or posteriorly. This pattern includes the previous term “patchy pattern without midline separation”. This pattern is a melanocytic/melanocyte precursor pattern as it is seen with mosaicism affecting melanocytes (can be increased or decreased pigmentation), and can also be seen with vascular mosaics.
Figure 1 – Three broad groups of recurrent developmental patterns on the skin affecting epidermal and melanocytic cell lineages, and sometimes including vascular lineages. Please see the text for the detailed description of these patterns. Importantly, cutaneous mosaicism is not confined to these patterns, and these patterns are not confined to cutaneous mosaicism.
Factors contributing to the incidence of mosaicism
At the bottom of table 1 there is an estimate of the comparative incidence of the different genetic mechanisms responsible for a mosaic abnormality presenting on the skin. There are several factors which contribute to the incidence of individual mosaic diseases. Before addressing these it is important to emphasise that the incidence of any mosaic is only an incidence at birth. Many mosaic mutations are lethal in early embryogenesis, and the timing cut off for whether a mutation is lethal or not likely varies from gene to gene and from cell lineage to cell lineage.
With that in mind the other factors which likely influence disease incidence at birth in mosaic disorders are:
- whether the disease phenotype only requires a single genetic hit – phenotypes caused by an autosomal dominant mutation affecting the skin will be more common than those which require two hits;
- whether the disease phenotype can be generated by mutations throughout the gene – for example mosaic disease-causing mutations in gene PIK3CA (producing PIK3CA-related overgrowth spectrum) have been described along its length whereas those in gene AKT1 (producing the AKT1-Proteus syndrome) are restricted to specific hotspots;
- the size of the disease gene - amongst those diseases where the phenotype can be generated by mutations in throughout the gene, diseases associated with larger genes are likely to be commoner as there are more bases to be mutated during cell division;
- the DNA sequence - amongst those diseases where the phenotype is generated by mutations in specific hotspots, it is likely that the DNA sequence itself will influence the incidence of that mutation, as we know from signatures of somatic mutagenesis15;
- it will be more common if the gene is very actively transcribed during embryogenesis in the cell type that produces the phenotype, or for universally actively transcribed genes during embryogenesis, as increased rates of transcription increase rates of somatic mutation16;
- it will be more common in certain families with predisposing or susceptibility variants in their germline genetics17,18.
Phenotypic variability in mosaic disease and its implications
Generally, we like genetic diseases to have clinical diagnostic criteria, for example those for neurofibromatosis type 1 (NF1), which help make a clinical diagnosis with a measurable degree of diagnostic certainty. We also like genetic diseases to have management guidelines, for example if the patient has an optic glioma they require regular checks for visual field defects. Neither of these standard practices are easy to achieve in mosaic disorders due to the enormous phenotypic variability within each disease, coupled to the rarity of the conditions. I strongly suspect that this is the reason for the sometimes enormous discrepancies in management recommendations in the literature in this field, and to frequent confusion amongst clinicians about what to do.
The phenotypic variability exists for two main reasons.
- The first of these is the timing of the mutation during pregnancy. Very early mutations will usually result in a higher proportion of the body cells carrying the mutation, and it will usually be present in more tissue types. This has been understood because exactly the same mutation has been found in differing severities of the same disease phenotype. For example, the same mutation can produce either an isolated portwine stain, or full blown Sturge-Weber syndrome (SWS) with skin, brain involvement19.
- The second source of major phenotypic variability is the lineage of the cell that is affected. A mutation in a vascular precursor will produce an entirely different disease phenotype than one in a melanocytic precursor. This has been understood because the same mutation has been found in entirely different mosaic diseases, for example BRAF p.(V600E) can cause arteriovenous malformations20 or linear syringocystadenoma papelliferum21. In fact, timing and lineage are also strongly interconnected, as eventually distinct cell lineages diverge originally from common precursors, and very early mutations may give rise to diseases with, for example, both vascular and melanocytic aspects.
As this new understanding has emerged, many previously distinct diseases are being re-grouped as spectra of diseases. For example in PIK3CA-related overgrowth spectrum (PROS) a large number of clinical diagnoses are now within this spectrum, and this has made it even more difficult to define diagnostic criteria.22 On the other hand there have been benefits for management, as the prescription of targeted therapies is now more likely to be driven by genotype than by phenotype, leading to PROS patients receiving PI3K or mTOR inhibitor treatment. The result of all of this is that most people in mosaics are now using a genotype-phenotype combined diagnosis, and it has recently been formally proposed by Biesecker in the context of Proteus syndrome.23
Genotype-Phenotype combined diagnostic terms
This is a simple concept. If both a clinical diagnosis can be made, and a genetic diagnosis can be made, the two are combined. Examples would be NRAS-Congenital melanocytic naevus syndrome, or MAP2K1-arteriovenous malformation. This type of diagnosis is understandable to patients whilst imparting the important genetic information to other physicians to help guide therapy. Where the mosaic disease presentation does not match any recognized clinical diagnosis, or the diagnosis is unknown to the clinician, the genetic diagnosis can be used alone, for example NRAS-mosaicism. Similarly, if a genetic diagnosis cannot be established – there are many genes still to be found – then the clinical diagnosis can be used alone.
How to reach a clinically-useful diagnosis of a patient presenting with a developmental pattern on the skin (figure 2)
- When you establish a clinical diagnosis or clinical diagnostic grouping, refer the patient to a patient support group. As with all rare disease where cures are lacking this is a very helpful intervention. Due to the uniqueness of phenotypes in mosaic disorders however there are often no appropriate support groups, and in those cases individual psychological support is imperative.
- Consider potential extra-cutaneous associations and investigate and refer appropriately. As a rough guide in mosaic conditions, if there are no guidelines/recommendations published from large cohorts about how to investigate have a high index of suspicion if there are symptoms or signs in other systems.
- Establish if the condition has a germline component or whether it is only mosaic. If there is a family history of the same developmental pattern it is extremely likely to be germline, even if you don’t know what the disease is (for example a linear epidermal naevus in a mother and daughter could be CHILD syndrome). The only other explanation would be that a rare mosaic disease has arisen independently in a parent and a child. If there is no family history it could be either germline or mosaic, unless it is a disease which is only ever mosaic (e.g. NRAS-congenital melanocytic naevus syndrome, or AKT1-Proteus syndrome, where the causative mutations are lethal in the germline). If it is potentially germline, or with a germline component, investigate the blood first for a causative genotype.
- If no germline component, biopsy the skin. To find the causative mutation within the cutaneous mosaicism you will very likely need to investigate the skin. Blood DNA or cheek swab DNA will almost never give you the answer.
- If biopsying the skin for a genotype, do not culture the skin cells. This will frequently lose or miss the mutation, because what most labs culture is fibroblasts. High quality DNA should be extracted directly from a fresh skin biopsy and sequenced deeply to look for mutations. If you are lucky enough to have a lab that can culture keratinocytes, melanocytes or endothelial cells, and you are very confident of the cell lineage affected, you can culture those as an alternative. If you are looking for an unknown/new gene, you will need blood DNA as a comparator to any skin sample.
- When you establish a genotype check if it has been described in the germline and refer appropriately for counselling. For conditions where a particular genotype has never been described in the germline (e.g. NRAS-congenital melanocytic naevus syndrome or AKT1-Proteus syndrome or GNAQ-Sturge-Weber syndrome) you can be confident that it will not be passed on in the germline to future offspring. If the genotype has been described in the germline (for example the patient has a FGFR2-epidermal naevus and the same mutation has been described in the germline causing severe thanatophoric dysplasia), refer the patient/family for genetic counselling even if passing on has never been documented in the literature. It is becoming increasing clear that where the germline heterozygous condition is not phenotypically-similar to the mosaic form (as in this example, unlike for example with KRT10 mutations where there is a clear phenotypic connection) it is likely that no-one has previously considered that these things could be connected and would therefore not have published.
- When you establish a genotype, if there is a clinical need for treatment check for trials of targeted therapies. As always not all patients will require therapy, and given the side effect profile of many targeted therapies these may not be appropriate. However, this field is moving so quickly that it is worth checking online for trials of targeted therapies, and considering the pros and cons of these in the context of the individual patient. These usually have inclusion criteria based on genotype.
Figure 2 – Overview of how to reach a clinically-useful diagnosis in a patient with a developmental pattern on the skin, and how to proceed.

References
1. Kinsler, V. A. et al. Mosaic abnormalities of the skin - review and guidelines from the European Reference Network for rare skin diseases (ERN-Skin). The British journal of dermatology, doi:10.1111/bjd.17924 (2019).
2. Paller, A. S. et al. Genetic and clinical mosaicism in a type of epidermal nevus. The New England journal of medicine 331, 1408-1415, doi:10.1056/NEJM199411243312103 (1994).
3. Weinstein, L. S. et al. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. The New England journal of medicine 325, 1688-1695, doi:10.1056/NEJM199112123252403 (1991).
4. Ju, Y. S. et al. Somatic mutations reveal asymmetric cellular dynamics in the early human embryo. Nature 543, 714-718, doi:10.1038/nature21703 (2017).
5. Martincorena, I. & Campbell, P. J. Somatic mutation in cancer and normal cells. Science 349, 1483-1489, doi:10.1126/science.aab4082 (2015).
6. Happle, R. Mosaicism in human skin. (Springer-Verlag, 2014).
7. Soblet, J. et al. Blue Rubber Bleb Nevus (BRBN) Syndrome Is Caused by Somatic TEK (TIE2) Mutations. The Journal of investigative dermatology 137, 207-216, doi:10.1016/j.jid.2016.07.034 (2017).
8. Vazquez-Osorio, I. et al. A case of mosaicism in ectodermal dysplasia-skin fragility syndrome. The British journal of dermatology 177, e101-e102, doi:10.1111/bjd.15374 (2017).
9. Happle, R. Mosaicism in human skin. Understanding the patterns and mechanisms. Archives of dermatology 129, 1460-1470 (1993).
10. Blaschko, A. in Bericht erstattet dem VII. Congress der Deutschen Dermatologischen Gesellschaft (Breslau, 1901).
11. Lehalle, D. et al. De novo mutations in the X-linked TFE3 gene cause intellectual disability with pigmentary mosaicism and storage disorder-like features. Journal of medical genetics 57, 808-819, doi:10.1136/jmedgenet-2019-106508 (2020).
12. Zweier, C. et al. A new face of Borjeson-Forssman-Lehmann syndrome? De novo mutations in PHF6 in seven females with a distinct phenotype. Journal of medical genetics 50, 838-847, doi:10.1136/jmedgenet-2013-101918 (2013).
13. Waelchli, R. et al. New vascular classification of port-wine stains: improving prediction of Sturge-Weber risk. The British journal of dermatology 171, 861-867, doi:10.1111/bjd.13203 (2014).
14. Kinsler, V. A. & Larue, L. The patterns of birthmarks suggest a novel population of melanocyte precursors arising around the time of gastrulation. Pigment cell & melanoma research 31(1), 95-109, doi:10.1111/pcmr.12645 (2018).
15. Alexandrov, L. B., Nik-Zainal, S., Wedge, D. C., Campbell, P. J. & Stratton, M. R. Deciphering signatures of mutational processes operative in human cancer. Cell Rep 3, 246-259, doi:10.1016/j.celrep.2012.12.008 (2013).
16. Jinks-Robertson, S. & Bhagwat, A. S. Transcription-associated mutagenesis. Annu Rev Genet 48, 341-359, doi:10.1146/annurev-genet-120213-092015 (2014).
17. Kinsler, V. A. et al. Germline melanocortin-1-receptor genotype is associated with severity of cutaneous phenotype in congenital melanocytic nevi: a role for MC1R in human fetal development. The Journal of investigative dermatology 132, 2026-2032, doi:10.1038/jid.2012.95 (2012).
18. Satyamaanasa Polubothu, L. A.-O., Daniël Lionarons, Mark Harland, et al. Germline duplications of PPP2R3B predispose to melanocytic naevi and melanoma by promoting a proliferative phenotype driven by C21orf91. Under review, Nature Communications June 2019, Online Preprint http://biorxiv.org/cgi/content/short/672576v1 (2019).
19. Shirley, M. D. et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. The New England journal of medicine 368, 1971-1979, doi:10.1056/NEJMoa1213507 (2013).
20. Al-Olabi, L. et al. Mosaic RAS/MAPK variants cause sporadic vascular malformations which respond to targeted therapy. J Clin Invest, doi:10.1172/JCI98589 (2018).
21. Levinsohn, J. L., Sugarman, J. L., Bilguvar, K., McNiff, J. M. & Choate, K. A. Somatic V600E BRAF Mutation in Linear and Sporadic Syringocystadenoma Papilliferum. The Journal of investigative dermatology, doi:10.1038/jid.2015.180 (2015).
22. Keppler-Noreuil, K. M. et al. PIK3CA-related overgrowth spectrum (PROS): diagnostic and testing eligibility criteria, differential diagnosis, and evaluation. American journal of medical genetics. Part A 167A, 287-295, doi:10.1002/ajmg.a.36836 (2015).
23. Sapp, J. C., Buser, A., Burton-Akright, J., Keppler-Noreuil, K. M. & Biesecker, L. G. A dyadic genotype-phenotype approach to diagnostic criteria for Proteus syndrome. Am J Med Genet C Semin Med Genet 181, 565-570, doi:10.1002/ajmg.c.31744 (2019).

Prof. Veronica Kinsler
MA MB BChir FRCPCH PhD
Professor of Paediatric Dermatology and Dermatogenetics at Great Ormond Street Hospital for Children and UCL GOS Institute of Child Health