
Kindler's Epidermolysis Bullosa
DISEASE CARD
| Disease group | Epithelial adhesion disorders |
|---|---|
| DISEASE NAME | KINDLER*S EPIDERMOLYSIS BULLOSA |
| Synonymous | - |
| Estimated prevalence | Unknown |
| OMIM | 173650 |
| Inheritance | Autosomal recessive |
| Gene (s) | FERMT1 (607900) |
Definition
Kindler epidermolysis bullosa (KEB), inherited autosomal recessive with mutations in FERMT1, is a rare subtype of epidermolysis bullosa with around 250 reported cases so far. It is characterized by congenital skin blistering, mild photosensitivity and progressive poikiloderma with diffuse skin atrophy.1, 2
Authors: Prof. Johann Bauer, MD Christine Prodinger
Clinical Description
KEB is characterized by congenital skin blistering (mostly acral and trauma-induced), similar to other forms of epidermolysis bullosa, with a propensity for mitigation with age in many patients, although the skin remains fragile throughout the life. Another typical symptom is photosensitivity that shows a variable degree of seveity (ranging from mild (facial burning/erythema) to severe (blister formation)) after minimal UV exposure and usually start to weakens during adolescence. At the age of 1-2 years of life, atrophy becomes evident usually on the dorsal aspects of hands and feet, then manifests on sun-exposed areas, before progressively spreading, eventually resulting in poikiloderma (areas of hyper-/hypopigmentation, reticular erythema, teleangiectasias) of the whole body at the end of the first decade of life.
The presence of palmoplantar keratoderma and nail abnormalities is variable. Frequently dermatoglyphics are lost. Occasionally, webbing of the fingers and pseudosyndactyly develops. Mucosal manifestations include gingival fragility, hemorrhagic gingivitis, labial leukokeratosis, poor dentition and, typically, early and rapidly progressive periodontitis. Other mucous membranes, i.e. urethral, anal, oesophageal and genital mucosae are affected less frequently, although severe manifestations (e.g. oeasophageal/urethral/vaginal stenosis/strictures, hemorrhagic diarrhea, ulcerative colitis) have been reported. Keratoconjunctivitis, ectropion and conjunctival scarring are associated eye pathologies. Squamous cell carcinomas (SCC), a feared complication in severe EB types, as the course is usually aggressive with a high rate of metastasis, have been reported as complications of KEB as well. In absence of SCC, life expectancy is normal.3-5
Pathogenesis
KEB is caused by mutations in the FERMT1 (syn. KIND1) gene localized in the short arm of chromosome 20. The kindlin protein is an intracellular protein, associated with integrins and focal adhesions. Histology of skin shows epidermal atrophy, focal vacuolization of basal keratinocytes and pigmentary incontinence in the upper dermis. Electron microscopy shows reduplication of the epidermal basement membrane and the different levels of blister formation.3, 6, 7
Diagnosis
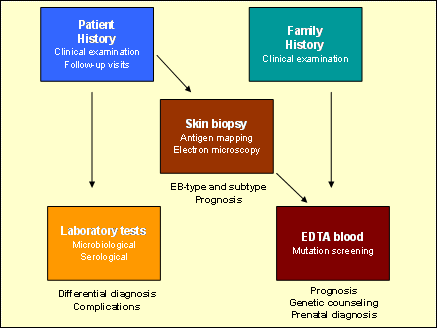
A detailed clinical skin examination is a precondition for diagnosis, although symptoms can mimic other subtypes of EB. The morphological analysis of a skin biopsy is the essential step in the diagnostic algorithm. These analyses reveal the level of the skin split that is in KEB mixed (i.e. either in the basal keratinocytes, in the lamina lucida or below the lamina densa). Antigen mapping demonstrates abnormal and discontinuous staining patterns of laminin 332 (previously laminin 5), α6-, ß4-integrin and collagens XVII and VII. Microbiological and serological tests may be needed to exclude primary or secondary infections and autoimmune diseases as a cause for skin blistering. Mutation detection is reasonable to confirm the diagnosis and essential for genetic counseling and prenatal diagnosis.8,7
Figure 1. Diagnostic algorithm for epithelial adhesion disorders (modified form www.netzwerk-eb.de)
Treatment
There is no causal therapy up-to-date. Patient care requires multidisciplinary management and collaboration of i.a. pediatricians, dermatologists, gastroenterologists, dentists, plastic surgeons, nutritionists and psychologists. Symptomatic therapy comprises a high standard of personal hygiene and daily skin care, protection from trauma and avoidance of infections. (see Treatment in the article "Epidermolysis bullosa") Blisters should be burst with a sterile needle, antiseptics and nonadhesive wound dressing are recommended for erosions/wounds. Patients should be educated about strict sun protection. Regular dental assessment and intensive care with oral hygiene is important for prevention and early treatment of periodontitis. Routine skin and mucosal examinations are inevitable for early detection of SCCs in adolescence and adulthood.
References
1. Has C, Bauer JW, Bodemer C, et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. 2020;183(4):614-627.

Prof. Johann Bauer, MBA
Professor of Dermatology
University Hospital of the Paracelsus Medical University Salzburg, Austria
Head of the EB Research Unit, EB House Austria, Salzburg